Definizione ed eziologia
Il rene policistico autosomico dominante (Autosomal dominant polycystic kidney disease - ADPKD) è la patologia renale genetica più comune con un'incidenza di 1/1000 ed è responsabile di circa il 5-10% dei pazienti con insufficienza renale terminale che necessitano di terapia sostitutiva. Le
manifestazioni cliniche sono rare prima dell'età adulta, ma la penetranza è pressochè completa: tutti i pazienti con età ≥ 80 anni hanno qualche segno.
Nell'86-96% dei casi, il rene policistico autosomico dominante è causata da mutazioni nel gene PKD1 sul cromosoma 16, che codifica per la proteina policistina 1; i restanti casi sono dovuti a mutazioni nel gene PKD2 sul cromosoma 4, che codifica per policistina 2. Pochi casi familiari non sono associati ad alcuno dei due loci.
Fisiopatologia
La policistina 1 regola l'adesione e la differenziazione delle cellule epiteliali tubulari.
La policistina 2 funge da canale ionico, le cui mutazioni determinano la secrezione di liquidi nelle cisti.
Le mutazioni di queste proteine possono alterare la funzione delle ciglia renali, che consentono alle cellule tubulari di essere sensibili alla velocità di flusso (apparentemente la proliferazione e la differenziazione delle cellule tubulari sono legate alla velocità di flusso e la disfunzione ciliare può pertanto causarne la trasformazione cistica).
Nelle fasi precoci della malattia, i tubuli si dilatano e lentamente si riempiono di filtrato glomerulare. Alla fine, i tubuli si separano dal nefrone funzionante e si riempiono di liquidi, formando le cisti. Si può verificare un'emorragia intracistica, che causa ematuria.
I pazienti sono anche a maggior rischio di pielonefrite acuta, infezione di cisti, e calcoli urinari (nel 20%).
Attraverso meccanismi ancora in fase di studio, si sviluppano sclerosi vascolare e fibrosi interstiziale che interessano circa il 10% dei tubuli; ciononostante, l’insufficienza renale si sviluppa in circa il 35-45% dei pazienti entro i 60 anni.
Manifestazioni extrarenali
- Altre manifestazioni cistiche
- Epatiche: le più frequenti
- Spleniche
- Pancreatiche
- Vescicole seminali
- Ovariche
- Aracnoidee
- Aneurisma intracranico (ICA)
- Presenti in circa il 4% dei giovani adulti e fino al 10% dei pazienti anziani
- Carcinoma a cellule renali
- Difetti valvolari cardiaci
- Prolasso della mitrale
- Insufficienza aortica: deriva dalla dilatazione della radice aortica dovuta ad alterazioni della parete arteriosa (tra cui aneurisma dell'aorta).
Clinica
Nelle prime fasi il rene policistico autosomico dominante non causa sintomi; la metà dei pazienti rimane asintomatica, non sviluppa mai insufficienza renale e la patologia non viene mai diagnosticata. Negli altri casi, grazie all'iper-filtrazione compensatoria, la sintomatologia si manifesta non prima della III/IV decade di vita.
Il decorso della patologia è caratterizzato dalla formazione progressiva di cisti renali a contenuto fluido e incremento del volume renale, responsabili del quadro clinico caratterizzato da:
- Dolore lombare o al fianco
- Ipertensione
- Ematuria
- Infezioni delle vie urinarie e delle cisti renali
- Insufficienza renale
Il dolore acuto può essere causato da
- Crescita cistica
- Infezione delle vie urinarie
- Nefrolitiasi
- Emorragia cistica
- Infezione cistica
L'anemia è meno frequente che in altri tipi di insufficienza renale cronica, presumibilmente perché la produzione di eritropoietina è conservata.
Quando la malattia è in fase avanzata, i reni possono aumentare massivamente di volume ed essere palpabili, determinando una sensazione di ripienezza nella parte superiore dell'addome e al fianco.
Diagnosi
La diagnosi viene posta sulla base dei seguenti esami:
- Anamnesi ed esame obiettivo
- Ipertensione
- Dolore lombare o al fianco
- Alterazione della funzionalità renale (non sempre presente)
- Ecografia
- Pazienti con storia familiare suggestiva e PKD1 mutato o genotipo sconosciuto (Criteri KDIGO):
- ≥ 3 cisti uni/bilaterali tra 15-39 anni
- ≥ 2 cisti uni/bilaterali tra 40-59 anni
- Ecografia negativa: valore predittivo negativo del 90-100%, sufficiente ad escludere la patologia nei pazienti tra 15-59 anni
- Pazienti con storia familiare negativa (necessaria conferma genetica):
- Ingrandimento renale e cisti
- Cisti epatiche
- Pazienti con storia familiare suggestiva e PKD1 mutato o genotipo sconosciuto (Criteri KDIGO):
- Genetica
- Utilizzata per la conferma diagnostica in casi selezionati
Imaging
Ecografia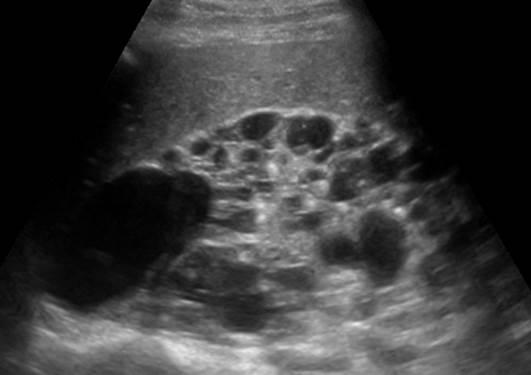
- Utile nella diagnosi secondo i Criteri KDIGO
- Permette di identificare:
- Ostruzioni delle vie escretrici urinarie
- Litiasi
- Cisti fino 2-3 mm
- Limiti
- Diagnosi in pazienti a rischio con età < 40 anni (indicata la valutazione con RM)
- Non permette di discriminare con accuratezza tra infezione o emorragia intracistica
TC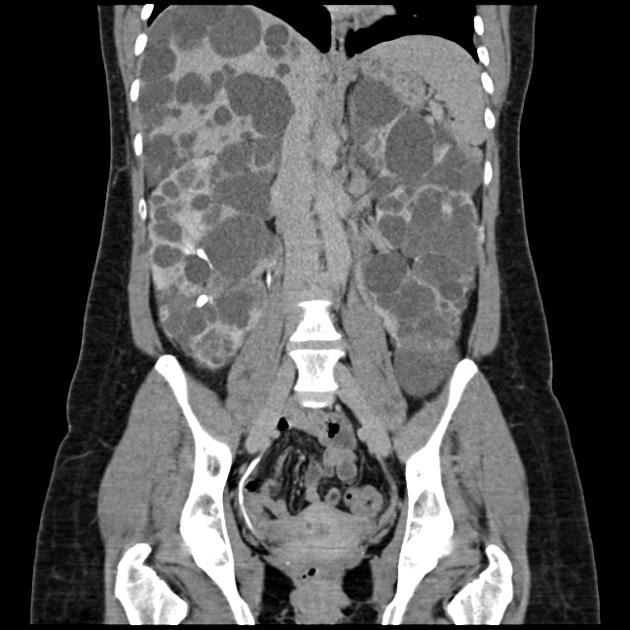
- Permette di identificare
- Litiasi
- Cisti emorragica
- Cisti infetta
- Calcolo del Total Kidney Volume (KTV)
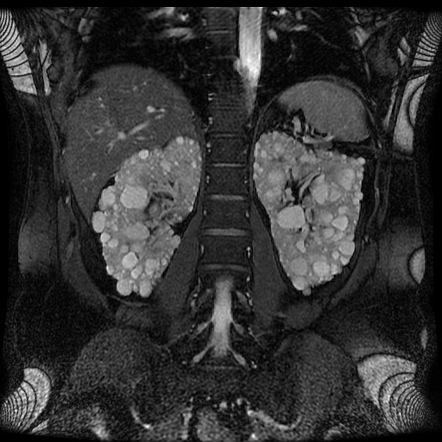
RM
- Indicata nella diagnosi dei pazienti tra 15-39 anni con storia familiare di ADPKD
- Diagnosi di ADPKD se > 10 cisti
- Esclusione di ADPKD se < 5 cisti
- Permette di distinguere tra cisti emorragiche e cisti infette
- Screening dell'Aneurisma IntraCranico (ICA)
- Secondo la KDIGO (Kidney Disease Improving Global Outcomes) non è raccomandato lo screening a tutta la popolazione affetta da ADPKD
- Lo screening si effettua con Angiografia RM senza mdc con tecnica Time-of-Flight (TOF)
- Le indicazioni allo screening sono:
- Storia familiare di aneurisma intracranico o emorragia subaracnoidea
- Sintomi suggestivi di ICA
- Precedente rottura di aneurisma intracranico
- Elevato rischio occupazionale (ad es. pilota di aereo)
- Valutazione pre-operatoria per interventi di chirurgia maggiore
- Misura del Total Kidney Volume (TKV)
- Il TKV è un indicatore dell'impatto (burden) delle cisti e della loro crescita
- La crescita annuale del TKV è di circa il 5-6%
- Può essere utilizzato per predire la progressione di malattia
- Associato al GRF può identificare i pazienti a rischio di insufficienza renale cronica
- Il rischio viene calcolato utilizzando un programma sviluppato dalla Mayo Clinic, disponibile a questo link
- Il TKV viene calcolato misurando, sugli assi propri del rene, le dimensioni massime renali sul piano coronale, sagittale e assiale
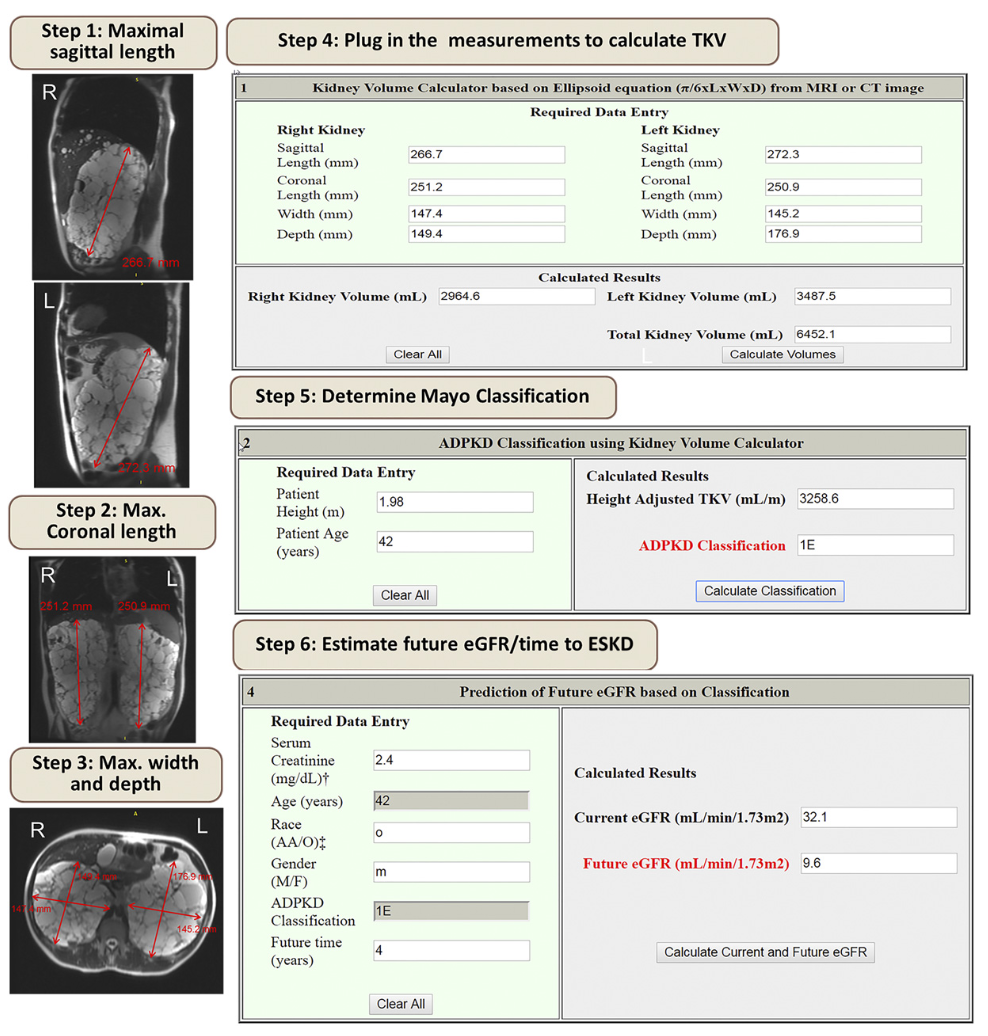
Classificazione Mayo Clinic
E' una classificazione basata sull'imaging.
- Classe 1: Tipica
- Distribuzione bilaterale e diffusa con sostituzione lieve/moderata/severa del parenchima renale da cisti, le quali contribuiscono tutte allo stesso modo al TKV (non si riconoscono sostanziali asimmetrie)
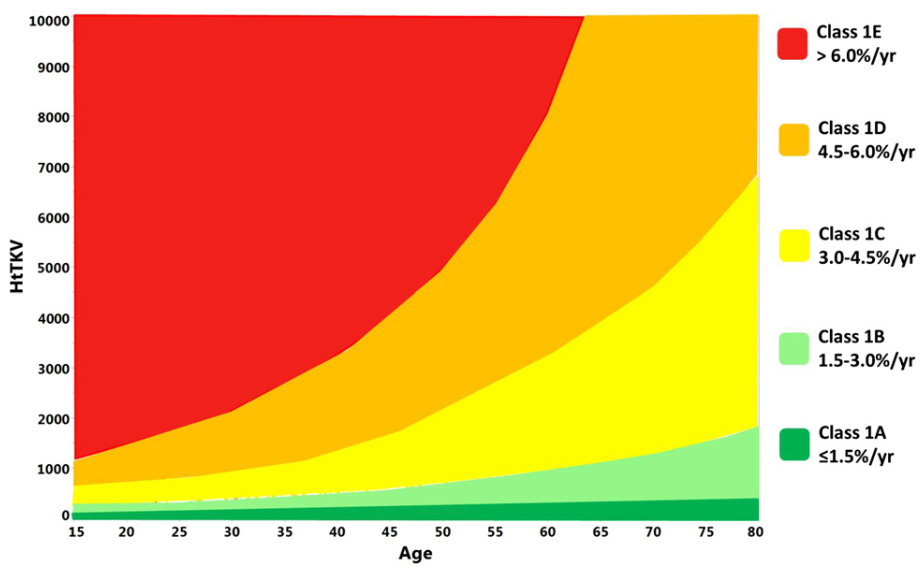
- Sottoclassi (ad ogni sottoclasse corrisponde una percentuale di crescita di TKV/anno):
- Classe 1A: basso rischio, non evolve a IRC
- Classe 1B: malattia lentamente progressiva, richiesto monitoraggio
- Classe 1C: malattia rapidamente progressiva, indicato il trattamento
- Classe 1D: malattia rapidamente progressiva, indicato il trattamento
- Classe 1E: malattia rapidamente progressiva, indicato il trattamento
- Classe 2: Atipica
- Classe 2A
- Unilaterale
- Segmentale
- Asimmetrica
- Lopsided
- Classe 2B
- Presentazione bilaterale con atrofia acquisita unilaterale
- Presentazione bilaterale con atrofia acquisita bilaterale
- Classe 2A
Fonti
-
Dynamed - Autosomal Dominant Polycystic Kidney Disease (ADPKD)
- Rene policistico autosomico dominante, E.Fung, MSD Manual
- KDIGO
- Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease, Fouad T. Chebib and Vicente E. Torres
- Radiopaedia
Raccolta di immagini
puoi provare a cercare con un'altra parola chiave oppure consultare l'Atlante di Radnote
 Atlante
Atlante